


It has currently been accepted that the interaction between environmental factors, and that of certain genes, can influence the destructive immune response characterized in many autoimmune diseases. As a matter of fact, approximately less than 10 percent of those people with a higher genetic susceptibility to disease may actually develop autoimmunity. This implies a solid environmental cause behind the beginning of the autoimmune process. Environmental factors have also been believed to likely affect the results of the process as well as the rate of development of autoimmune diseases. One theory is that intestinal luminal antigens absorbed through the gut might be involved in the pathogenesis of autoimmune diseases. The intestinal epithelium is the largest mucosal surface in the human body and it provides a connection between the external environment and the mammalian host.
Table of Contents
What environmental factors cause autoimmune diseases?
Healthy, mature intestinal mucosa with its absolute tight junctions, or TJs, is the most significant barrier for the passage of macromolecules, as seen on Figure 1. In a physiological state, quantitatively small but immunologically active antigens can cross the mucosal barrier. These antigens are absorbed through the mucosa via two practical paths. The massive collection of absorbed proteins, amounting to about 90 percent, cross the intestinal barrier throughout the transcellular pathway followed by lysosomal degradation which converts the proteins into smaller, non-immunogenic peptides. The remaining proteins are then carried as entire proteins, causing antigen-specific immune responses in the body. This occurrence utilizes the Microfold (M) cell pathway or the paracellular pathway, which requires a subtle but complex balance of intercellular TJs that can result in antigenic tolerance.
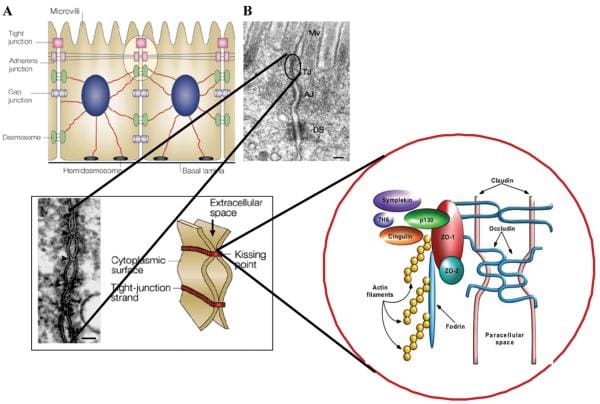
After the integrity of the intestinal barrier are compromised, best known as TJ disassembly, an immune response to environmental antigens that spanned the gut mucosa can grow, leading to autoimmune diseases or allergies. The cells that play a vital part in this immune response lie in close proximity to the intestinal epithelial barrier. Another critical component for this immune response is the human leukocyte antigen, or HLA, system. HLA class I and II genes encode the antigen presenting cell (APC) glycoprotein receptors that present antigens to T cells in the intestinal mucosa. Susceptibility to up to 50 diseases, such as celiac disease, or CD, and type 1 diabetes, or T1D, has been associated with certain HLA class I or class II alleles. A typical denominator of these diseases is the occurrence of numerous preexisting conditions which can lead to autoimmunity. The first is a hereditary susceptibility for the host immune system to recognize, and potentially misinterpret, an environmental antigen introduced within the gastrointestinal tract, or GI tract. Second, the host needs to be exposed to the antigen. Finally, the antigen needs to be introduced into the gastrointestinal mucosal immune system, following its M-cell passage or paracellular passage, usually blocked by TJ competency, from the intestinal lumen to acquire the intestine submucosa. In most instances, higher intestinal permeability precedes disease and triggers an abnormality in antigen delivery which triggers an immune response, ultimately causing autoimmunity. Researchers have therefore hypothesized that genes, environment, and decreased intestinal barrier function are all critical to develop autoimmune diseases, especially CD and T1D.
Gliadin as an Environmental Factor of Autoimmune Diseases
Celiac Disease
Gluten is a well-known environmental factor that triggers celiac disease. It is the gliadin fraction of wheat germ and equal alcohol-soluble proteins in distinct grains, known as prolamins, which are connected to the growth of intestinal damage. A standard characteristic of the prolamins of wheat, rye, and barley is a greater content of glutamine (>30%) and proline (>15%), whereas the non-toxic prolamins of rice and corn have decreased glutamine and proline content. However, the environmental factor that influenced the development CD is complex and unknown. Some aspects of gluten consumption might help determine the risk of CD incidence, particularly in: the amount of gluten intake, the higher the amount, the larger the risk; the caliber of consumed gluten, a few grains contain more hazardous epitopes than others; and the pattern/timing of infant feeding. Recent research studies suggest that the pattern of infant nutrition might have a very important role on the development of the CD as well as that of other autoimmune diseases. Breastfeeding is believed to delay or reduce the possibility of developing CD. The positive effects of breast milk may be attributed to its influence on the microbial colonization procedure for the own newborn’s intestine. The genus Bifidobacterium is predominant in the feces of breast-fed infants, while a larger variety of bacterial groups, including Bacteroides, Streptococcus, Clostridium, etc., are found in the fecal microbiota of all formula-fed infants. Changes in the composition of the intestinal microbiota also occur as a consequence of the following changes from breastfeeding or formula feeding to weaning and even the introduction of solid food. Alterations in the intestinal balance between favorable and possibly harmful bacteria have also been associated with allergy symptoms, type 1 diabetes and inflammatory bowel diseases, among others.
Type 1 Diabetes
It is believed that genetically predisposed individuals develop T1D after encountering one or more environmental factors of the disease. Fast improvements could be made in disease prevention and treatment if these environmental factors were identified. Amongst the others, gliadin has only been the subject of a series of research studies that aim at establishing its part in the pathogenesis of type 1 diabetes. Early introduction of gliadin-containing cereals were reported to raise the prospect of islet cell autoimmunity in humans. Gliadin-specific, lamina propria-derived T cells play an important role in the pathogenesis of CD. The same HLA class II haplotype, DQ (? 1 * 0501, ß1 * 0201), that can be connected with gliadin peptides in CD is also one of two HLA class II haplotypes inherited most frequently by people with T1D. There are also signs of immunological activity in the intestine of T1D patients: jejunal specimens from T1D patients have been found to consist of much higher doses of interferon gamma (IFN?)- and tumor necrosis factor-alpha (TNF-?) positive cells in contrast to people with healthy controls, suggesting an inflammatory response. Still another study found substantially increased manifestation of HLA-DR and HLA-DP molecules on intestinal villi of jejunal specimens from T1D patients in comparison with specimens from healthy controls. Recent evidence confirmed these findings by assessing the mucosal immune response to gliadin in the jejunum of patients with T1D. Small intestinal biopsies from children with T1D were cultured with gliadin and evaluated for epithelial infiltration and lamina propria T-cell activation. The caliber of intraepithelial CD3+ cells and of lamina propria CD25+ mononuclear cells has been higher in jejunal biopsies from T1D patients versus control subjects. In the patients’ biopsies cultured with enzymatically treated gliadin, there was epithelial infiltration by CD3 cells, a more significant growth in lamina propria CD25+ and CD80+ cells, enhanced manifestation of lamina propria cells favorable into ligand and receptor molecules ?4/?7 and ICAM 1, along with enhanced expression of CD54 and crypt HLA-DR. Also, ?4 positive T cells have been recovered in the pancreatic islets of an T1D person, providing an immediate connection between gliadin-activated T cells and destruction of pancreatic islet cells.
Findings from research studies using non-obese diabetic, or NOD, mice in addition to the BioBreeding diabetes-prone, or BBDP, rats have also implicated wheat gliadin as a nutritional supplement diabetogen. In BBDP rats, gliadin vulnerability is accompanied by increased intestinal permeability, and changes in gut microbiota composition, as seen on Figure 2., presumably allow food antigens to grow in contact with all the underlying lamina propria. Feeding NOD mice and BBDP rats a gluten free hydrolyzed casein diet resulted in a delay and decline in T1D development. Interestingly, these T1D animal models additionally demonstrated the moment of exposure to wheat proteins is quite important to the development of T1D. Delaying the vulnerability of diabetogenic wheat proteins by prolonging the breastfeeding period decreased T1D expansion from the BBDP rats. What is more, exposing neonatal rats or mice to diabetogenic wheat components or bacterial antigens diminished T1D incidence, which is perhaps due to the induction of immunological tolerance.
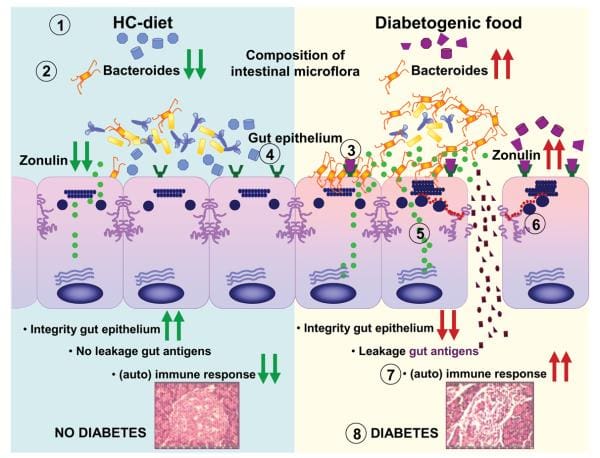
Rats that were fed corn protein-based diets developed T1D and demonstrated a moderate celiac-like enteropathy. Mesenteric lymph nodes, or MLNs, which drain the gut, are the substantial inductive site where dietary antigens are famous in the gut-associated connective tissue. The authors described an increase in the expression ratio of T-bet:Gata3, master transcription factors for Th1 and Th2 cytokines, respectively, in the MLN by wheat-fed BBDP rats compared to this by BBDR rats, mainly due to diminished Gata3 expression. Also, CD3+CD4+IFN?+ T cells were prevalent in the MLN of wheat-fed BBDP rats, but remained at control levels in BBDP rats fed with a diabetes-retardant wheat-free diet. BioBreeding diabetes-prone MLN cells increased quickly in response to wheat protein antigens in a particular, dose-dependent manner, and 93 percent of cells were CD3+CD4+ T cells. This proliferation was connected using a minimum proportion of CD4+CD25+ T cells and a greater proportion of dendritic cells in the MLN of BBDP rats. These results suggest that, before insulitis is established, the MLNs of wheat-fed BBDP rats contain a remarkably large proportion of Th1 cells that rapidly increased particularly in response to wheat protein antigens. Collectively, these research studies suggest a deranged mucosal immune response to gliadin in T1D and a direct connection between gliadin-induced stimulation of gut mucosal T cells and abuse of pancreatic islet cells, as seen on Figure 2.
Link between Gliadin, Zonulin & Increased Intestinal Permeability in Autoimmune Diseases
Researchers have generated enough evidence to support that gliadin can induce increased intestinal permeability by releasing preformed zonulin. Intestinal cell lines exposed to gliadin released zonulin from the cell medium with subsequent zonulin binding to the cell surface, rearrangement of the cell cytoskeleton, loss of occludin-ZO1 protein interaction, and increased monolayer permeability. Pre-treatment with all of the zonulin antagonist AT1001 blocked these alterations without affecting zonulin release. When exposed to luminal gliadin, intestinal biopsies from patients with celiac disease in remission expressed a continuous luminal zonulin discharge and increase in intestinal permeability. On the contrary, biopsies from non-CD patients showed a limited, transient zonulin release, which was paralleled by a decline in intestinal permeability that had not reached the level of permeability found in celiac disease cells. As a matter of fact, when gliadin was added to the basolateral side of cell lines or intestinal biopsies, no zonulin release was detected. The latter finding indicates that gliadin interacts using an intestinal luminal receptor, which encouraged researchers to comprehend this issue. In vitro experiments revealed specific colocalization of gliadin along with the chemokine receptor CXCR3 expressed in human and mouse intestinal epithelium and lamina propria. Gliadin vulnerability led to a tangible establishment of CXCR3 and MyD88. Ex vivo experiments revealed that gliadin exposure to intestinal segments from wild-type mice increased zonulin terminal and intestinal permeability, whereas CXCR3 intestinal segments failed to respond to gliadin. The increased intestinal permeability appeared cause a specific impact for gliadin, because the subsequent CXCR3 ligand, IP-10, did not affect intestinal barrier function. Based on these figures, researchers suggested that gliadin contrasts to CXCR3 additionally lead to stimulation of the zonulin pathway and improved intestinal permeability in a MyD88-dependent manner.
Conclusive Remarks
The classical paradigm of the pathogenesis of autoimmune diseases involving certain receptor makeup and exposure to environmental factors was contested with the addition of a third component, the decrease of intestinal barrier function. Genetic predisposition, miscommunication between innate and adaptive immunity, exposure to environmental factors and loss in intestinal barrier function secondary to the breakdown of intercellular tight junctions, or TJs, seem to be vital components in the pathogenesis of autoimmune disorders. Both in CD and T1D gliadin may play a role in inducing loss of intestinal barrier function or inducing the gastrointestinal response in genetically predisposed individuals. This new hypothesis suggests that after the digestive process is triggered, it is not auto-perpetuating, but rather, it might be balanced or reversed by preventing the continuous interaction between genes and the environment. Since TJ dysfunction allows this interaction, new treatment procedures targeted at re-establishing the intestinal barrier function supply innovative, unexplored procedures for caring for autoimmune diseases. Information referenced from the National Center for Biotechnology Information (NCBI) and the National University of Health Sciences. The scope of our information is limited to chiropractic and spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
By Dr. Alex Jimenez

Additional Topics: Wellness
Overall health and wellness are essential towards maintaining the proper mental and physical balance in the body. From eating a balanced nutrition as well as exercising and participating in physical activities, to sleeping a healthy amount of time on a regular basis, following the best health and wellness tips can ultimately help maintain overall well-being. Eating plenty of fruits and vegetables can go a long way towards helping people become healthy.

WELLNESS TOPIC: EXTRA EXTRA: Managing Workplace Stress
1. Fasano A. Tight Junctions. CRC Press, Inc.; Boca Raton, FL: 2001. Pathological and therapeutic implications of macromolecule passage through the tight junction; pp. 697–722.
2. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat. Rev. Immunol. 2003;3:331–341. [PubMed]
3. Fasano A. Intestinal zonulin: open sesame! Gut. 2001;49:159–162. [PMC free article] [PubMed]
4. Brandtzaeg P, Halstensen TS, Kett K, et al. Immunobiology and immunopathology of human gut mucosa: humoral immunity and intraepithelial lymphocytes. Gastroenterol. 1989;97:1562–1584. [PubMed]
5. Brandtzaeg P. Overview of the mucosal immune system. Curr. Top. Microbiol. Immunol. 1989;146:13–25. [PubMed]
6. Bjorkman PJ, Saper MA, Samraoui B, et al. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. [PubMed]
7. Bjorkman PJ, Saper MA, Samraoui B, et al. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:512–518. [PubMed]
8. Cuvelier C, Mielants H, De Vos M, et al. Major histocompatibility complex class II antigen (HLA-DR) expression by ileal epithelial cells in patients with seronegative spondylarthropathy. Gut. 1990;31:545–549.[PMC free article] [PubMed]
9. Wendling D. Role of the intestine in the physiopathology of inflammatory rheumatism. Rev. Rhum. Mal. Osteoartic. 1992;59:389–392. [PubMed]
10. Bjarnson I, Williams P, Smethurst P, et al. Effect of non-steroidal anti-inflammatory drugs and prostaglandins on the permeability of the human small intestine. Gut. 1986;27:1292–1297.[PMC free article] [PubMed]
11. Bjarnason I, Peters TJ, Levi AJ. Intestinal permeability: clinical correlates. Dig. Dis. 1986;4:83–92.[PubMed]
12. Pratesi R, Gandolfi L, Garcia SG, et al. Prevalence of coeliac disease: unexplained age-related variation in the same population. Scand. J. Gastroenterol. 2003;38:747–50. [PubMed]
13. Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study. Arch. Int. Med. 2003;163:286–292. [PubMed]
14. Nistico L, Fagnani C, Coto I, et al. Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut. 2006;55:803–808. [PMC free article] [PubMed]
15. Louka AS, Sollid LM. HLA in coeliac disease: unravelling the complex genetics of a complex disorder. Tissue Antigens. 2003;61:105–117. [PubMed]
16. Vader W, Stepniak D, Kooy Y, et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc. Natl. Acad. Sci. USA. 2003;100:12390–12395. [PMC free article] [PubMed]
17. Monsuur AJ, Bakker PI, Alidazeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat. Gen. 2005;37:1341–1344. [PubMed]
18. Wapenaar MC, Monsuur AJ, van Bodegraven AA, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–467. [PubMed]
19. Kelly MA, Rayner ML, Mijovic CH, et al. Molecular aspects of type 1 diabetes. Mol. Pathol. 2003;56:1–10. [PMC free article] [PubMed]
20. Santiago JL, Martinez A, Nunez C, et al. Association of MYO9B haplotype with type 1 diabetes. Hum. Immunol. 2008;69:112–115. [PubMed]
21. Sollid LM. Breast milk against celiac disease. Gut. 2002;51:767–768. [PMC free article] [PubMed]
22. Grönlund M-M, Arvilommi H, Kero P, et al. Importance of intestinal colonization in the maturation of humoral immunity in early infancy: a prospective follow up study of healthy infants aged 0–6 months. Arch. Dis. Child. Fetal. Neon. 2000;83:F186–F192. [PMC free article] [PubMed]
23. Kirjavainen PV, Arvola T, Salminen SJ, et al. Aberrant composition of gut microbiota of allergic infants: a target of bifidobacterial therapy at weaning? Gut. 2002;51:51–55. [PMC free article] [PubMed]
24. Sartor RB. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterol. 2004;126:1620–1633. [PubMed]
25. Lefebvre DE, Powell KL, Strom A, et al. Dietary proteins as environmental modifiers of type 1 diabetes mellitus. Annu. Rev. Nutr. 2006;26:175–202. [PubMed]
26. Ziegler AG, Schmid S, Huber D, et al. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA. 2003;290:1721–1728. [PubMed]
27. Norris JM, Barriga K, Klingensmith G, et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA. 2003;290:1713–1720. [PubMed]
28. Lundin KEA, Scott H, Hansen T, et al. Gliadin-specific, HLA-DQ (?180501,ß1 * 0201) restricted T cells isolated from the small intestinal mucosa of celiac patients. J. Exp. Med. 1993;178:187–196.[PMC free article] [PubMed]
29. Agardh D, Nilsson A, Tuomi T, et al. Prediction of silent celiac disease at diagnosis of childhood type 1 diabetes by tissue transglutaminase autoantibodies and HLA. Pediatric Diab. 2001;2:58–65. [PubMed]
30. Westerholm-Ormio M, Vaarala O, Pihkala P, et al. Imunologic activity in the small intestinal mucosa of pediatric patients with type 1 diabetes. Diabetes. 2003;52:2287–2295. [PubMed]
31. Savilahti E, Ormala T, Saukkonen U, et al. Jejuna of patients with insulin-dependent diabetes mellitus (IDDM) show signs of immune activation. Clin. Exp. Immunol. 1999;116:70–77. [PMC free article][PubMed]
32. Auricchio R, Paparo F, Maglio M, et al. In vitro deranged intestinal immune response to gliadin in type 1 diabetes. Diabetes. 2004;53:1680–1683. [PubMed]
33. Hanninen A, Salmi M, Simell O, et al. Endothelial cell-binding properties of lymphocytes infiltrated into human diabetic pancreas: Implications for pathogenesis in IDDM. Diabetes. 2003;42:1656–1662.[PubMed]
34. Chakir H, Lefebvre DE, Wang H, et al. Wheat protein-induced proinflammatory T helper 1 bias in mesenteric lymph nodes of young diabetes-prone rats. Diabetologia. 2005;48:1576–1584. [PubMed]
35. Scott FW, Cloutier HE, Kleeman R, et al. Potential mechanisms by which certain foods promote or inhibit the development of spontaneous diabetes in BB rats. Dose, timing, early effect on islet area, and switch in infiltrate from Th1 to Th2 cells. Diabetes. 1997;46:589–598. [PubMed]
36. Funda DP, Kaas A, Taskalova-Hogenova H, et al. Gluten-free but also gluten-enriched (gluten+) diet prevent diabetes in NOD mice; the gluten enigma in type 1 diabetes. Diab. Metab. Res. Rev. 2008;24:59–63. [PubMed]
37. Meddings JB, Jarand J, Urbanski SJ, et al. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am. J. Physiol. 1999;276:G951–957. [PubMed]
38. Visser J, Brugman S, Klatter F, et al. Short-term dietary adjustment with a hydrolyzed casein-based diet postpones diabetes development in the diabetes-prone BB rat. Metabolism. 2003;52:333–337. [PubMed]
39. Brugman S, Klatter F, Visser J, et al. Neonatal oral administration of DiaPep277, combined with hydrolysed casein diet, protects against Type 1 diabetes in BB-DP rats. An experimental study. Diabetologia. 2004;47:1331–1333. [PubMed]
40. Brugman S, Klatter F, Visser J, et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia. 2006;49:2105–2108. [PubMed]
41. Visser J, Groen H, Klatter F, et al. The diabetes prone BB rat model of IDDM shows duration of breastfeeding to influence Type 1 diabetes development later in life. Diabetologia. 2003;46:1711–1713.[PubMed]
42. Scott FW, Rowsell P, Wang GS, et al. Oral exposure to diabetes-promoting food or immunomodulators in neonates alters gut cytokines and diabetes. Diabetes. 2002;51:73–78. [PubMed]
43. Fasano A, Fiorentini C, Donelli G, et al. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J. Clin. Invest. 1995;96:710–720.[PMC free article] [PubMed]
44. Fasano A, Uzzau S, Fiore C, et al. The enterotoxic effect of zonula occludens toxin (Zot) on rabbit small intestine involves the paracellular pathway. Gastroenterol. 1997;112:839–846. [PubMed]
45. Marcial MA, Carlson SL, Madara JL. Partitioning of paracellular conductance along the ileal crypt-villus axis: a hypothesis based on structural analysis with detailed consideration of tight junction structure-function relationships. J. Membr. Biol. 1984;80:59–70. [PubMed]
46. Uzzau S, Lu R, Wang W, et al. Purification and preliminary characterization of the zonula occludens toxin receptor from human (CaCo2) and murine (IEC6) intestinal cell lines. FEMS Microbiol. Lett. 2001;194:1–5. [PubMed]
47. Wang W, Uzzau S, Goldblum SE, et al. Human zonulin, a potential modulator of intestinal tight junctions. J. Cell Sci. 2000;113:4435–4440. [PubMed]
48. Fasano A, Baudry B, Pumplin DW, et al. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. USA. 1991;88:5242–5246. [PMC free article] [PubMed]
49. Baudry B, Fasano A, Ketley JM, et al. Cloning of a gene (zot) encoding a new toxin produced by Vibrio cholerae. Infect. Immun. 1992;60:428–434. [PMC free article] [PubMed]
50. Di Pierro M, Lu R, Uzzau S, et al. Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J. Biol. Chem. 2001;276:19160–19165. [PubMed]
51. El Asmar R, Panigrahi P, Bamford P, et al. Host-dependent activation of the zonulin system is involved in the impairment of the gut barrier function following bacterial colonization. Gastroenterol. 2002;123:1607–1615.
52. Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;358:1518–1519. [PubMed]
53. Watts T, Berti I, Sapone A, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type-I diabetes in BB diabetic prone rats. Proc. Natl. Acad. Sci. USA. 2005;102:2916–2921. [PMC free article] [PubMed]
54. Sapone A, de Magistris L, Pietzak M, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. 2006;55:1443–1449. [PubMed]
55. Clemente MG, Virgiliis S, Kang JS, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. 2003;52:218–223. [PMC free article] [PubMed]
56. Drago S, El Asmar R, De Pierro M, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand. J. Gastroenterol. 2006;41:408–419. [PubMed]
57. Lammers KM, Lu R, Brownley J, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterol. 2008;135:194–204.[PMC free article] [PubMed]
58. Barbeau WE, Bassaganya-Riera J, Hontecillas R. Putting the pieces of the puzzle together – a series of hypotheses on the etiology and pathogenesis of type 1 diabetes. Med. Hypotheses. 2007;68:607–619.[PubMed]
Post Disclaimer
General Disclaimer, Licenses and Board Certifications *
Professional Scope of Practice *
The information herein on "Environmental Factors for Autoimmune Diseases" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and on our family practice-based chiromed.com site, focusing on naturally restoring health for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine; wellness; contributing etiological viscerosomatic disturbances within clinical presentations; associated somato-visceral reflex clinical dynamics; subluxation complexes; sensitive health issues; and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and licensure jurisdiction. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that directly or indirectly relate to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
New Mexico CNP License#: 90560, Verified
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
| Yes | 363LF0000X - Nurse Practitioner - Family | NM |
90560 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
📆 Schedule Appointment: Schedule 24/7 (Click Here)