


How often do you have a hard time remembering your appointments? Has it become harder for you to learn new things? How often do you feel you have something that must be done? Or even, how often do you feel more susceptible to pain? Research studies have demonstrated that brain fog may be associated with Alzheimer’s disease. In the following article, we will discuss how midlife systemic inflammatory markers have ultimately been associated with late-life brain volume.
Table of Contents
Midlife Systemic Inflammatory Markers are Associated with Late-life Brain Volume
Abstract
- Objective: To clarify the temporal relationship between systemic inflammation and neurodegeneration, we examined whether a higher level of circulating inflammatory markers during midlife was associated with smaller brain volumes in late-life using a large biracial prospective cohort study.
- Methods: Plasma levels of systemic inflammatory markers (fibrinogen, albumin, white blood cell count, von Willebrand factor, and Factor VIII) were assessed at baseline in 1,633 participants (mean age 53 [5] years, 60% female, 27% African American) enrolled in the Atherosclerosis Risk in Communities Study. Using all 5 inflammatory markers, an inflammation composite score was created for each participant. We assessed episodic memory and regional brain volumes, using 3T MRI, 24 years later.
- Results: Each SD increase in midlife inflammation composite score was associated with 1,788 mm3 greater ventricular (p = 0.013), 110 mm3 smaller hippocampal (p = 0.013), 519 mm3 smaller occipital (p = 0.009), and 532 mm3 smaller Alzheimer disease signature region (p = 0.008) volumes, and reduced episodic memory (p = 0.046) 24 years later. Compared to participants with no elevated (4th quartile) midlife inflammatory markers, participants with elevations in 3 or more markers had, on average, 5% smaller hippocampal and Alzheimer disease signature region volumes. The association between midlife inflammation and late-life brain volume was modified by age and race, whereby younger participants and white participants with higher levels of systemic inflammation during midlife were more likely to show reduced brain volumes subsequently.
- Conclusions: Our prospective findings provide evidence for what may be an early contributory role of systemic inflammation in neurodegeneration and cognitive aging.
Introduction
Although elevated levels of inflammatory markers have been found in the blood,1 CSF,2 and brain parenchyma3 of individuals with cognitive impairment and Alzheimer disease (AD), it remains unclear whether this heightened inflammatory state is driving neurodegenerative changes. If low-grade systemic inflammation does play a causal role in AD and other neurodegenerative diseases, a heightened inflammatory response during midlife would be expected to increase one’s risk for pathologic brain changes much later. Although cross-sectional studies have demonstrated a link between elevated inflammatory markers and reduced brain volume in older adults,4,–7 it remains unclear whether systemic inflammation during midlife, before the onset of significant age- and disease-related neurologic changes, is associated with brain volume loss later in life.
The goal of the current study was to examine how midlife plasma markers of inflammation relate to late-life brain volume among a biracial community sample of older adults. To this end, we examined the relationship between 5 markers of systemic inflammation measured during midlife and MRI measures of regional brain volume 24 years later in the Atherosclerosis Risk in Communities (ARIC) Study cohort. We tested the hypothesis that greater midlife systemic inflammation is associated with smaller brain volumes in regions most susceptible to AD-related atrophy and reduced episodic memory in older adulthood. Based on cross-sectional evidence suggesting that race, sex, and age may modify the association between inflammatory markers and brain volume,5,8,9 the current study also examined the modifying effects of each of these demographic characteristics.
Methods
Study population. The ARIC study, an ongoing community-based prospective study, enrolled 15,792 middle-aged adults (45–65 years of age at baseline).10 Participants were selected by probability sampling in 4 US communities: Washington County, Maryland; Forsyth County, North Carolina; northwestern suburbs of Minneapolis, Minnesota; and Jackson, Mississippi. Following the baseline visit in 1987–1989 (visit 1), participants were seen at 3 more visits, approximately 3 years apart until 1996–1998 (visit 4), and at the fifth visit in 2011–2013 (visit 5).
At visit 5, a subset of 1,978 participants was selected to undergo brain MRI scans.11 Participants were selected to undergo a brain MRI based on previous participation in the ARIC Brain MRI Ancillary Study and standard safety exclusion criteria. In addition, all participants with evidence of cognitive impairment at visit 5 and an age-stratified random sample of participants without evidence of cognitive impairment were recruited. The participation rate among eligible individuals selected to undergo brain MRI was approximately 81%. A detailed description of the MRI sampling strategy is provided in the e-Methods at Neurology.org. We excluded participants with poor imaging quality (n = 6), neurologic disease (i.e., stroke, multiple sclerosis) (n = 80), missing inflammatory biomarker data (n = 38), missing covariates (n = 215), and race other than white or African American (n = 6). Participants who met criteria for dementia (5%, n = 83) were excluded from the primary analyses.
Standard protocol approvals, registrations, and patient consents. The ARIC study protocol has been approved by the institutional review boards at each participating center. All participants gave written informed consent at each study visit.
Inflammatory markers. Plasma levels of 4 acute-phase reactants—fibrinogen, albumin, von Willebrand factor (VWF), and Factor VIII (FVIII)—and white blood cell (WBC) count were used to measure systemic inflammation.12 Using standard protocols, study technicians drew fasting blood, centrifuged samples, and froze plasma blood samples at ?70°C until the samples were analyzed.13 Fibrinogen (mg/dL), albumin (g/dL), VWF (% of standard), and FVIII activity (% of standard) measured at visit 1 were analyzed in an ARIC research laboratory in accordance with a standardized protocol.13,14 WBC count was determined from whole anticoagulated blood using an automated particle Coulter Counter within 24 hours of venipuncture. Repeated testing revealed interassay coefficients of variation below 8% for fibrinogen, albumin, FVIII, and WBC, and 17%–19% for VWF.15,16
Brain MRI. MRI scans were conducted using a 3T MRI scanner.11 Magnetization-prepared rapid gradient echo (MPRAGE), axial T2* gradient recalled echo, axial T2 fluid-attenuated inversion recovery, and axial diffusion tensor imaging sequences were obtained. Freesurfer (surfer.nmr.mgh.harvard.edu) was used to measure brain volume from MPRAGE sequences.17 Total brain and ventricular volume, lobar volume (frontal, temporal, parietal, occipital), AD signature region volume (i.e., the combined volume of the parahippocampal, entorhinal, inferior parietal lobules, hippocampus, and precuneus),18 hippocampal volume, and total intracranial volume were evaluated for the current study.
Episodic memory. Episodic memory was assessed at visit 5, concurrent with the brain MRI, using the delayed word recall test (DWR). DWR is a test that requires participants to learn and recall a list of 10 words following a delay period.19 Participants were scored based on the total number of words correctly recalled.
Covariates. Race, sex, years of education attained (less than high school, high school/General Equivalency Development/vocational school, or any college), cigarette smoking status (current/former/never), average weekly alcohol consumption (grams), and previous cancer diagnosis were self-reported. A random zero sphygmomanometer was used to calculate sitting diastolic and systolic blood pressure. Second and third blood pressure measurements were averaged for the current analyses. Hypertension was defined as systolic blood pressure >140 mm Hg, diastolic blood pressure >90 mm Hg or use of hypertensive medication. Body mass index was calculated using recorded height and weight (kg/m2). Coronary heart disease was defined as self-reported coronary bypass, balloon angioplasty, angioplasty of one or more coronary artery, or myocardial infarction. Medications used in the previous 2 weeks were recorded. The presence of chronic inflammatory conditions (e.g., arthritis, lupus, gout) was assessed by patient self-report of physician diagnosis at visit 4. History of regular anti-inflammatory medication use (e.g., nonsteroidal anti-inflammatory drug, arthritis medication) was assessed at visit 5. All other variables were assessed at visit 1. Dementia diagnosis was adjudicated at visit 5 by an expert committee using cognitive, imaging, and functional data.20
Total cholesterol and triglycerides were measured using enzymatic methods,21,22 and low-density lipoprotein using the Friedewald equation.23 Serum glucose was measured using the hexokinase method. Diabetes was defined as a fasting glucose ?126 mg/dL or a nonfasting glucose ?200 mg/dL, current use of diabetes medication or insulin, or participant report of physician-diagnosed diabetes. APOE genotype (0, 1, or 2 ?4 alleles) was assessed using the TaqMan assay (Applied Biosystems, Foster City, CA).
Statistical analysis. We examined systemic inflammation as both a continuous and categorical exposure measure. A continuous inflammation composite Z score was created using the 5 inflammatory markers. WBC count was log-transformed to correct for skewness. Each inflammatory biomarker was converted to a standardized Z score such that the group mean was zero with an SD of 1. The mean of the 5 Z scores was calculated to generate an inflammation composite Z score. Because albumin decreases in response to inflammation, albumin values were multiplied by ?1 before being included in the composite Z score. With few exceptions, the intercorrelations between inflammatory markers were within an optimal range, between 0.2 and 0.4; composite score item–test correlations, principal component factor loadings, and Cronbach ? (0.61) were satisfactory for our purposes (table e-1). For each participant, we also created a categorical measure of systemic inflammation by computing the number of inflammatory marker Z scores in the highest quartile (?75%tile) and trichotomizing this number (0, 1–2, or 3–5).
Participant characteristics were compared using an analysis of variance or ?2 tests. Multivariable linear regression was used to assess the association between continuous and categorical inflammation variables and measures of brain volume and episodic memory. Brain volume analyses were adjusted for total intracranial volume, and all analyses included the covariates described in the previous section. Interaction terms or stratification were used to evaluate the modifying effects of age, race, and sex.
Sensitivity analyses were performed excluding participants who reported regular anti-inflammatory medication use during follow-up and including participants who met criteria for dementia. For all analyses, sampling weights were incorporated to account for the ARIC brain MRI sampling strategy. Thus, all results represent estimates for the entire ARIC visit 5 study population. Because the associations between inflammation markers and specific regions of interest (ROIs) are correlated, we did not adjust for multiple comparisons. A 2-sided p-value <0.05 designated statistical significance. All analyses were conducted using Stata Version 14 (StataCorp, College Station, TX).
Results
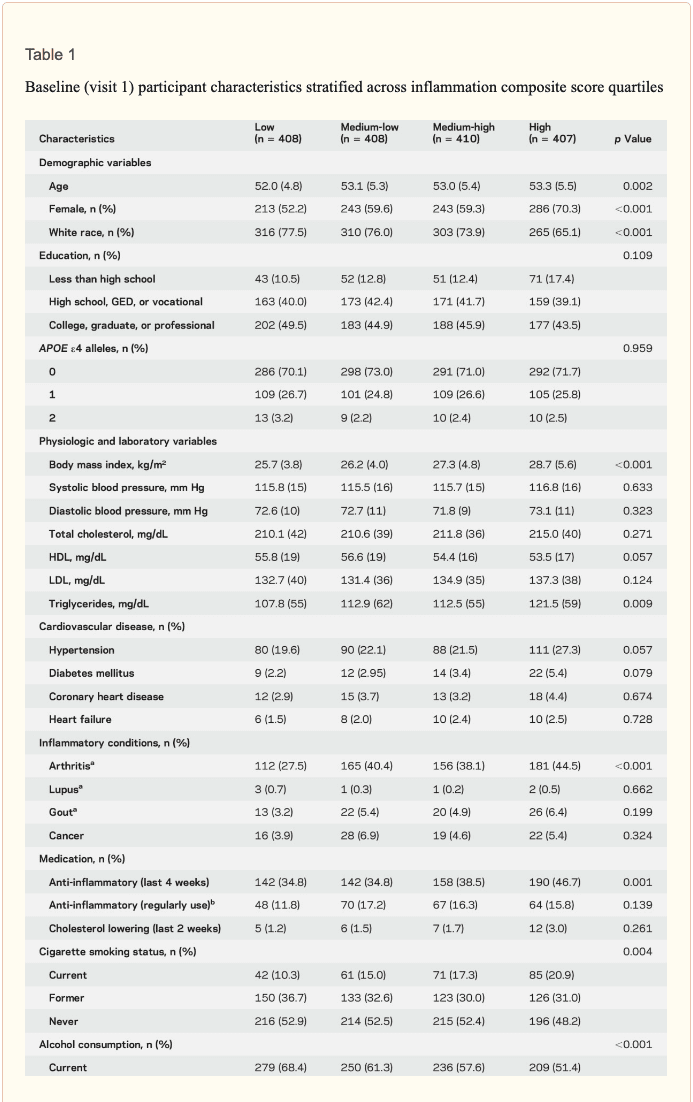
Study population characteristics. A total of 1,633 participants (baseline mean age 52.8 [5.3] years, 27% African American, 60% women, 46% college or professional degree) were included in the study sample. The time between baseline assessment and follow-up MRI scan was 24 (1) years; the average age at follow-up was 76.5 (5.4) years. As shown in table 1, a higher inflammation composite score at baseline was associated with older age, female sex, African American race, and increased levels of a number of cardiovascular risk factors.
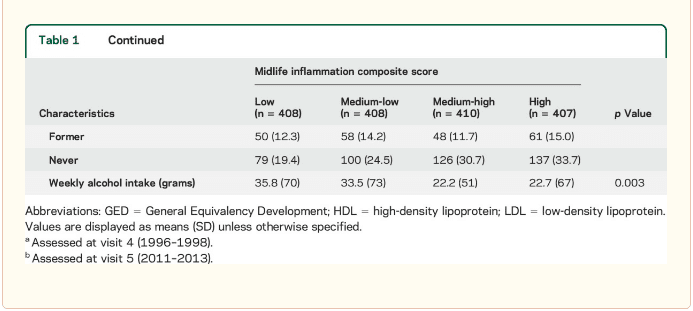
Inflammatory markers and brain volume. Each SD increase in inflammation composite score at baseline was associated with a 532 mm3 smaller AD signature region volume (95% confidence interval [CI] ?922 to ?141), a 519 mm3 smaller occipital lobe volume (CI ?906 to ?132), a 110 mm3 smaller hippocampal volume (CI ?196 to ?24), and a 1,788 mm3 larger ventricular volume (CI 371 to 3,205) at follow-up (table 2). We found the estimated effect of a 1 SD increase in inflammation composite score during midlife on occipital lobe, ventricular, and hippocampal volume to be similar to the effect associated with possession of a single APOE ?4 allele in our multivariable regression analyses. No association was found for the total brain, frontal lobe, temporal lobe, or parietal lobe volume (ps > 0.071). Our findings did not change meaningfully after excluding participants who regularly used anti-inflammatory medication during the follow-up period (table e-2) and after including participants who met the criteria for dementia at visit 5 (table e-3). For descriptive purposes, associations between individual inflammatory markers and AD signature region volume are provided in a table e-4.
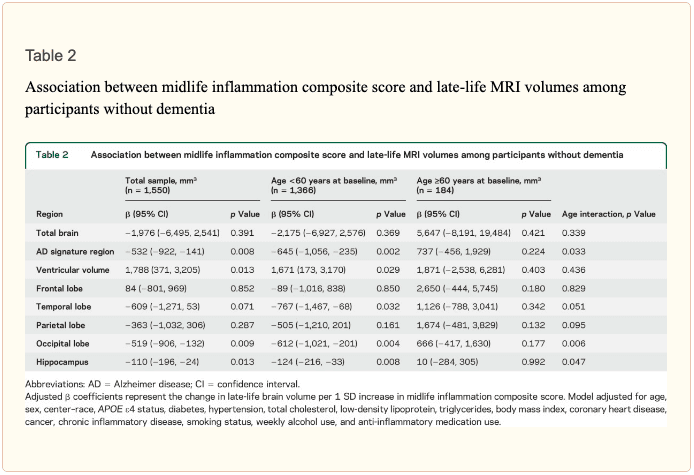
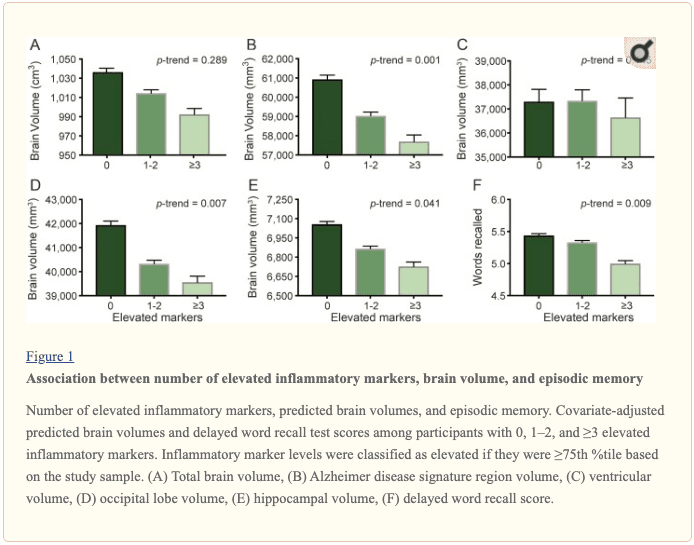
An assessment of linear trend revealed that compared to individuals with 0 elevated (?75th %tile) inflammatory biomarkers at baseline (reference), those with 1–2 and 3–5 elevated biomarkers had lower AD signature region (p trend = 0.001), occipital lobe (p trend = 0.007), and hippocampal volume (p trend = 0.041) 24 years later (figure 1). Compared to the reference group, participants with 3 or more elevated markers demonstrated 5.3% smaller AD signature region volumes, 5.7% smaller occipital lobe volumes, and 4.6% smaller hippocampal volumes, on average. However, this pattern was not statistically supported for the total brain, ventricular, frontal lobe, temporal lobe, and parietal lobe volume (p trends >0.072).
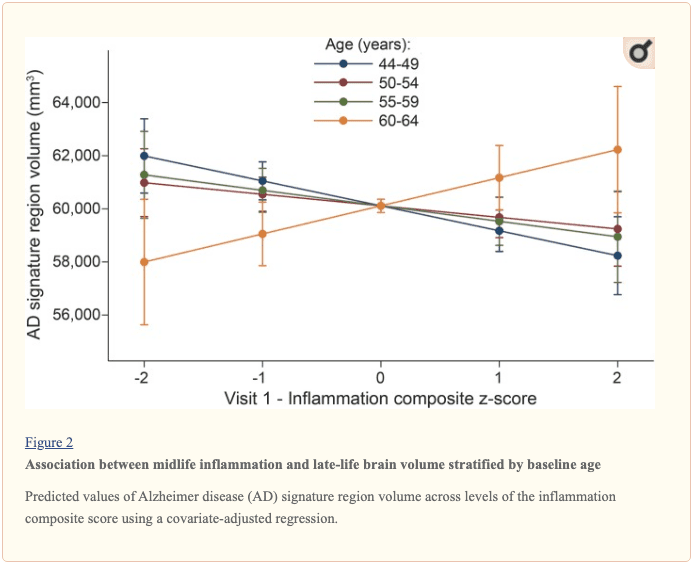
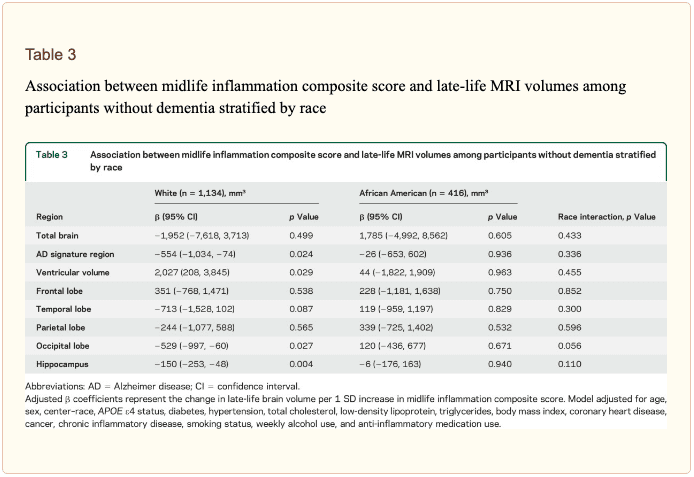
The modifying effects of age, race, and sex. A significant age-by-inflammation composite score interaction was found for the AD signature region, occipital lobe, and hippocampal volume (table 2). Because a reversal of association was observed at age 60 (figures 2, e-1, and e-2), we stratified the sample into young-midlife and old-midlife subgroups (<60/? 60). As displayed in table 2, the associations between higher midlife inflammation composite score and lower AD signature region, occipital lobe, and hippocampal volume at follow-up were significantly stronger among participants who were 60 or younger at baseline compared to those who were older than 60. A marginal race-by-inflammation composite score interaction was found for occipital lobe volume, whereby a higher midlife inflammation composite score was associated with lower occipital lobe volume among white, but not African American, participants (table 3). No interactions with sex were found (table e-5).
Inflammatory markers and episodic memory. Late-life episodic memory, which was associated with hippocampal and AD signature region volume after controlling for age (partial rs > 0.21, ps < 0.001), was reduced among participants with higher levels of the inflammation composite score. Each SD increase in inflammation composite score was associated with a ?0.08 SD performance decrement on the DWR after adjusting for covariates (CI ?0.15 to 0.00; p = 0.046). Similarly, a higher number of elevated inflammatory biomarkers at baseline was associated with reduced DWR performance (p trend = 0.009; figure 1).
Discussion
Using a large community sample, we demonstrated that a higher level of systemic inflammatory markers measured during midlife is independently associated with lower regional brain volume and reduced episodic memory 24 years later among older adults without dementia. Similarly, participants who had elevations in a larger number of 5 inflammatory markers during midlife were found to have lower regional brain volumes and reduced episodic memory in late-life in a dose-response manner. For several brain regions, including the hippocampus, the effect of a 1 SD increase in midlife inflammation composite score was comparable to that of possessing a single APOE ?4 allele during late life. Whereas age and race were found to modestly modify the relationship between midlife inflammation and late-life regional brain volume, the previously reported modifying effect of sex was supported.
Although cross-sectional evidence from the Framingham5 study and several other population-based8,9 studies suggests an association between brain volume and inflammation in older adults, the temporal relationship between inflammation and brain volume loss is still not well-understood. As a result, whether heightened systemic inflammation constitutes a potential cause or consequence of neurodegeneration and brain atrophy remains unclear. Because the pathophysiologic processes driving neurodegeneration and brain volume loss begin decades before the onset of frank cognitive decline,24 it is essential to determine how biological processes that take place during middle adulthood relate to neurologic outcomes later in life. By demonstrating that an elevation in plasma inflammatory markers during midlife is independently associated with smaller regional brain volumes, larger ventricular volume, and reduced episodic memory in late life, the current findings provide support for a potential causal, rather than associative, role of systemic inflammation in late-life neurodegeneration (i.e., atrophy) and resulting cognitive decline. The current findings align closely with those from the neurocardiovascular literature, which have found associations between midlife blood pressure,25 cholesterol,26 and diabetes27 and adverse neurologic and cognitive outcomes in older adulthood. The contributing role of systemic inflammation to subsequent neurodegenerative processes has been demonstrated previously by animal studies,28 but had not yet been supported by a large prospective MRI study.
The current results suggest that several demographic factors modify the relationship between midlife inflammation and late-life brain volume. Younger individuals with elevated levels of inflammation (particularly participants in their 40s) were more likely to display lower brain volumes decades later, supporting the idea that elevated systemic inflammation earlier in life may make individuals especially vulnerable to neurodegenerative brain changes as they age. Although we expected stronger effects would emerge within the African American group, given the greater burden of systemic disease29 and dementia,30 the associations between inflammation and brain volume were generally weaker among African Americans. A previous study that examined the moderating effects of race found similar results in a cross-sectional analysis of older adults without dementia.8
Circulating levels of acute-phase reactants, such as those used in the current study, change in parallel with an inflammatory response as a result of signaling from inflammatory cytokines such as interleukin-6 and tumor necrosis factor-?.12 Cytokines in the periphery have the potential to induce a pro-inflammatory neurotoxic state within the CNS through multiple routes, including activation of endothelial cells of the blood-brain barrier,31 activation of macrophage in circumventricular organs,32 and signaling of the afferent vagus nerve.33 In addition to providing support for a pathogenic role of systemic inflammation in neurodegenerative disease, the present findings indicate that elevations in commonly assayed inflammatory proteins may serve as markers of risk for future neurodegenerative changes and cognitive decline. Although we did not examine all brain regions in our analysis, our assessment of 7 representative ROIs suggests that brain regions vulnerable to atrophy, amyloid deposition, and metabolic abnormalities in the earliest phases of AD may be more vulnerable to volume loss associated with heightened midlife inflammation. This pattern of neuroanatomic specificity has been supported by previous cross-sectional studies of older adults without dementia.4,7,–9,34
In the context of the current findings, several alternative explanations should be considered. First, it remains possible that elevated systemic inflammation may simply serve as a marker of another pathologic process linked to neurodegeneration (e.g., oxidative stress). Second, it is possible that the biological processes causing brain atrophy to trigger a protective neuroimmune response, which increases peripheral inflammation. Third, the associations found here may be an effect of residual or unmeasured confounding. Despite these caveats, the contributory role of systemic inflammation has been supported by a sizable body of literature implicating peripheral inflammatory signaling in neurodegenerative processes such as neural apoptosis,35 ?-amyloid formation,36 and neuronal tau phosphorylation.37
Strengths of the current study include the prospective study design, length of follow-up, detailed assessment of potentially confounding variables, large sample size, and the inclusion of a large African American sample. However, the current findings should be interpreted within the context of several limitations. Although the acute-phase reactants used in the present study represent components of the innate immune system, several of these proteins are implicated in another closely related physiologic process, such as hemostasis, which may also influence brain volume. Evaluating inflammatory biomarkers that have greater biological specificity in future prospective studies will allow for stronger inferences about the contributing role of systemic inflammation. Interpretation of the current findings is also limited by the measurement of inflammatory markers at a single time point, as it is unclear whether a single measurement can adequately capture inflammation chronicity. The relatively high interassay variability of VWF also increases the likelihood of exposure misclassification; however, this possibility is mitigated by the use of the inflammation composite score. We found that participants who dropped out and participants who died before visit 5 had significantly higher levels of midlife inflammation, were older, had greater levels of medical comorbidity at baseline, and were more likely to be African American38 (table e-6). As a result, selective attrition may have biased results in the direction of the null hypothesis, particularly for African American and older participants. Finally, our interpretation of the contributory role of inflammation in neurodegeneration rests on the assumption that brain volume loss occurred after inflammatory markers were assessed. Although evidence suggests that this is likely the case (brain volume loss accelerates after age 60 years39), this cannot be confirmed without the assessment of change over time.
Despite these limitations, the current study provides insights into the connection between midlife systemic inflammation and late-life brain volume loss. These findings provide support for inflammation’s early pathogenic role in the development of neurodegenerative brain changes associated with late-life cognitive decline, AD, and other forms of dementia.

Is inflammation the final trip wire for Alzheimer’s disease? Research studies have demonstrated that neuroinflammation is considered to be the main epigenetic trip wire for the genetic predisposition of Alzheimer’s disease or AD. Moreover, patients with inflammation can also develop a variety of symptoms, including brain fog which can make thinking, understanding, and remembering basic information challenging. Neuroinflammation can cause brain fog and other other well-known health issues, including Alzheimer’s disease and other neurological diseases. – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Neurotransmitter Assessment Form
The following Neurotransmitter Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptoms listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
In honor of Governor Abbott’s proclamation, October is Chiropractic Health Month. Learn more about the proposal.
Have you been experiencing noticeable variations in your mental speed? Do you suffer from pain, discomfort, and inflammation? Have you been experiencing fatigue, especially after meals or exposure to chemicals, scents, or pollutants? Brain fog can cause a variety of symptoms, including memory and concentration as well as vision problems. According to the research study above, midlife inflammation and brain fog may be associated with Alzheimer’s disease.
The following article has been referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues or functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
1. Tan ZS, Beiser AS, Vasan RS, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham study. Neurology 2007;68:1902–1908. [PubMed] [Google Scholar]
2. DeKosky ST, Ikonomovic MD, Wang X, et al. Plasma and cerebrospinal fluid alpha1-antichymotrypsin levels in Alzheimer’s disease: correlation with cognitive impairment. Ann Neurol 2003;53:81–90. [PubMed] [Google Scholar]
3. Minett T, Classey J, Matthews FE, et al. Microglial immunophenotype in dementia with Alzheimer’s pathology. J Neuroinflammation 2016;13:135. [PMC free article] [PubMed] [Google Scholar]
4. Zhang H, Sachdev PS, Wen W, et al. The relationship between inflammatory markers and voxel-based gray matter volumes in nondemented older adults. Neurobiol Aging 2016;37:138–146. [PubMed] [Google Scholar]
5. Jefferson AL, Massaro JM, Wolf PA, et al. Inflammatory biomarkers are associated with total brain volume: the Framingham Heart Study 43. Neurology 2007;68:1032–1038. [PMC free article] [PubMed] [Google Scholar]
6. Wersching H, Duning T, Lohmann H, et al. Serum C-reactive protein is linked to cerebral microstructural integrity and cognitive function. Neurology 2010;74:1022–1029. [PubMed] [Google Scholar]
7. Bettcher BM, Wilheim R, Rigby T, et al. C-reactive protein is related to memory and medial temporal brain volume in older adults. Brain Behav Immun 2012;26:103–108. [PMC free article] [PubMed] [Google Scholar]
8. Schmidt MF, Freeman KB, Windham BG, et al. Associations between serum inflammatory markers and hippocampal volume in a community sample. J Am Geriatr Soc 2016;64:1823–1829. [PMC free article] [PubMed] [Google Scholar]
9. Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3C-Dijon Study. Neurology 2012;78:720–727. [PubMed] [Google Scholar]
10. Hill C, Gerardo D, James F, et al. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives: The ARIC Investigators. Am J Epidemiol 1989;129:687–702. [PubMed] [Google Scholar]
11. Knopman DS, Griswold ME, Lirette ST, et al. Vascular imaging abnormalities and cognition: mediation by cortical volume in nondemented individuals: Atherosclerosis Risk in Communities: neurocognitive study. Stroke 2015;46:433–440. [PMC free article] [PubMed] [Google Scholar]
12. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 1999;340:448–454. [PubMed] [Google Scholar]
13. Papp AC, Hatzakis H, Bracey A, Wu KK. ARIC hemostasis study: I: development of a blood collection and processing system suitable for multicenter hemostatic studies. Thromb Haemost 1989;61:15–19. [PubMed] [Google Scholar]
14. Folsom AR, Wu KK, Rosamond WD, Sharrett AR, Chambless LE. Prospective study of hemostatic factors and incidence of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation 1997;96:1102–1108. [PubMed] [Google Scholar]
15. Chambless LE, McMahon R, Wu K, Folsom A, Finch A, Shen YL. Short-term intraindividual variability in hemostasis factors: The ARIC study: Atherosclerosis Risk in Communities intraindividual variability study. Ann Epidemiol 1992;2:723–733. [PubMed] [Google Scholar]
16. Eckfeldt JH, Chambless LE, Shen YL. Short-term, within-person variability in clinical chemistry test results: experience from the Atherosclerosis Risk in Communities Study. Arch Pathol Lab Med 1994;118:496–500. [PubMed] [Google Scholar]
17. Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [PubMed] [Google Scholar]
18. Dickerson BC, Stoub TR, Shah RC, et al. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology 2011;76:1395–1402. [PMC free article] [PubMed] [Google Scholar]
19. Schneider ALC, Sharrett AR, Gottesman RF, et al. Normative data for 8 Neuropsychological tests in older blacks and whites from the Atherosclerosis Risk in Communities (ARIC) study. Alzheimer Dis Assoc Disord 2015;29:32–44. [PMC free article] [PubMed] [Google Scholar]
20. Knopman DS, Gottesman RF, Sharrett AR, et al. Mild cognitive impairment and dementia prevalence: the Atherosclerosis Risk in Communities neurocognitive study: Alzheimer’s dementia diagnosis. Assess Dis Monit 2016;2:1–11. [PMC free article] [PubMed] [Google Scholar]
21. Nägele U, Hägele EO, Sauer G, et al. Reagent for the enzymatic determination of serum total triglycerides with improved lipolytic efficiency. Clin Chem Lab Med 1984;22:165–174. [PubMed] [Google Scholar]
22. Siedel J, Hagele EO, Ziegenhorn J, Wahlefeld AW. Reagent for the enzymatic determination of serum total cholesterol with improved lipolytic efficiency. Clin Chem 1983;29:1075–1080. [PubMed] [Google Scholar]
23. McNamara JR, Cohn JS, Wilson PWF, Schaefer EJ. Calculated values for low-density lipoprotein cholesterol in the assessment of lipid abnormalities and coronary disease risk. Clin Chem 1990;36:36–42. [PubMed] [Google Scholar]
24. Chetelat G, Baron JC. Early diagnosis of Alzheimer’s disease: contribution of structural neuroimaging. Neuroimage 2003;18:525–541. [PubMed] [Google Scholar]
25. Gottesman RF, Schneider ALC, Albert M, et al. Midlife hypertension and 20-year cognitive change. JAMA Neurol 2014;71:1218–1227. [PMC free article] [PubMed] [Google Scholar]
26. Kivipelto M, Helkala EL, Hänninen T, et al. Midlife vascular risk factors and late-life mild cognitive impairment: a population-based study. Neurology 2001;56:1683–1689. [PubMed] [Google Scholar]
27. Rawlings AM, Sharrett AR, Schneider ALC, et al. Diabetes in midlife and cognitive change over 20 years: a cohort study. Ann Intern Med 2014;161:785–793. [PMC free article] [PubMed] [Google Scholar]
28. Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol 2013;9:25–34. [PubMed] [Google Scholar]
29. Mensah GA, Mokdad AH, Ford ES, Greenlund KJ, Croft JB. State of disparities in cardiovascular health in the United States. Circulation 2005;111:1233–1241. [PubMed] [Google Scholar]
30. Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. J Am Med Assoc 2002;287:329–336. [PubMed] [Google Scholar]
31. Jurgens HA, Johnson RW. Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp Neurol 2012;233:40–48. [PMC free article] [PubMed] [Google Scholar]
32. Lacroix S, Feinstein D, Rivest S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol 1998;8:625–640. [PubMed] [Google Scholar]
33. Johnston GR, Webster NR. Cytokines and the immunomodulatory function of the vagus nerve. Br J Anaesth 2009;102:453–462. [PubMed] [Google Scholar]
34. Baune BT, Konrad C, Grotegerd D, et al. Tumor necrosis factor gene variation predicts hippocampus volume in healthy individuals. Biol Psychiatry 2012;72:655–662. [PubMed] [Google Scholar]
35. Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005;25:9275–9284. [PMC free article] [PubMed] [Google Scholar]
36. Sastre M, Walter J, Gentleman SSM, et al. Interactions between APP secretases and inflammatory mediators. J Neuroinflammation 2008;5:25. [PMC free article] [PubMed] [Google Scholar]
37. Krstic D, Madhusudan A, Doehner J, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation 2012;9:151. [PMC free article] [PubMed] [Google Scholar]
38. Gottesman RF, Rawlings AM, Sharrett AR, et al. Impact of differential attrition on the association of education with cognitive change over 20 years of follow-up. Am J Epidemiol 2014;179:956–966. [PMC free article] [PubMed] [Google Scholar]
39. Hedman AM, van Haren NEM, Schnack HG, Kahn RS, Hulshoff Pol HE. Human brain changes across the life span: a review of 56 longitudinal magnetic resonance imaging studies. Hum Brain Mapp 2012;33:1987–2002. [PubMed] [Google Scholar]
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease

Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual’s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Formulas for Methylation Support

XYMOGEN’s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
![]()
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All of the above XYMOGEN policies remain strictly in force.
Post Disclaimer
Professional Scope of Practice *
The information herein on "Functional Neurology: Midlife Brain Fog and Alzheimer's Disease?" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Our information scope is limited to Chiropractic, musculoskeletal, acupuncture, physical medicines, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somatovisceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and/or functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for the injuries or disorders of the musculoskeletal system.
Our videos, posts, topics, subjects, and insights cover clinical matters, issues, and topics that relate to and directly or indirectly support our clinical scope of practice.*
Our office has reasonably attempted to provide supportive citations and has identified the relevant research studies supporting our posts. We provide copies of supporting research studies available to regulatory boards and the public upon request.
We understand that we cover matters that require an additional explanation of how it may assist in a particular care plan or treatment protocol; therefore, to further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez, DC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, RN*, CCST, IFMCP*, CIFM*, ATN*
email: coach@elpasofunctionalmedicine.com
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License # TX5807, New Mexico DC License # NM-DC2182
Licensed as a Registered Nurse (RN*) in Florida
Florida License RN License # RN9617241 (Control No. 3558029)
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Presently Matriculated: ICHS: MSN* FNP (Family Nurse Practitioner Program)
Dr. Alex Jimenez DC, MSACP, RN* CIFM*, IFMCP*, ATN*, CCST
My Digital Business Card
 Again We Welcome You¸
Again We Welcome You¸
